Reviewed by the Help Dementia Editorial Team — our editors review every article for accuracy against guidance from the National Institute on Aging, the Alzheimer’s Association, and peer-reviewed sources.
Late dementia sits at the center of this dementia and brain health question.
LATE—limbic-predominant age-related TDP-43 encephalopathy—mimics Alzheimer’s disease in its most recognizable symptom: progressive memory loss. Both diseases destroy a person’s ability to remember new information and events, both strike primarily in the oldest age groups, and both result in visible brain atrophy on imaging. But beneath this surface similarity lies a fundamental biological difference. LATE is caused not by the amyloid-beta and tau proteins that hallmark Alzheimer’s disease, but by accumulation of a protein called TDP-43—and this difference makes all the difference when deciding how to treat it. A person diagnosed with pure Alzheimer’s disease might benefit from medications like lecanemab that clear amyloid from the brain; that same medication would do nothing for someone with pure LATE because the underlying pathology is completely different. This article examines why LATE so convincingly masquerades as Alzheimer’s, what distinguishes it at the biological level, and why the treatment paradigm is shifting away from a one-size-fits-all dementia approach toward precision medicine that targets the actual pathology driving each patient’s decline.
The stakes for accurate diagnosis are high. LATE is not rare. Among people aged 85 and older—the fastest-growing segment of the U.S. population—approximately one in three has LATE. Many people actually have both Alzheimer’s and LATE simultaneously, a co-pathology that accelerates cognitive decline faster than either disease alone. Doctors have only recently developed reliable clinical tools to tell them apart. This guide walks through the clinical clues that point to LATE, the emerging treatments that specifically target TDP-43, and what the 2025 diagnostic criteria mean for patients and families navigating dementia care.
Table of Contents
- Why Does LATE Look So Much Like Alzheimer’s Disease?
- The Biological Distinction—TDP-43 vs. Amyloid-Beta and Tau
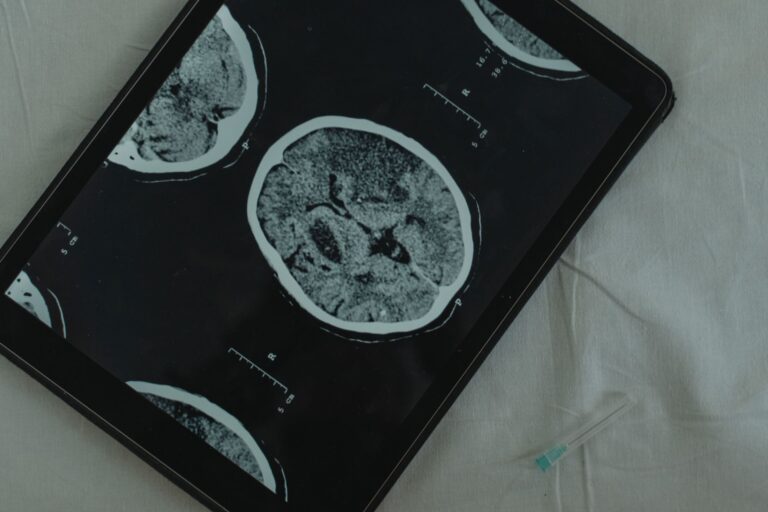
- Medial Temporal Lobe Atrophy—The Imaging Clue
- Why Current Alzheimer’s Medications Don’t Work for LATE
- Gene Therapy and Precision TDP-43 Treatments on the Horizon
- The 2025 Diagnostic Criteria—How Doctors Now Reliably Identify LATE
- The Prevalence Epidemic—Why LATE Is Far More Common Than Most People Realize
- Conclusion
Why Does LATE Look So Much Like Alzheimer’s Disease?
Both LATE and Alzheimer’s disease present with progressive episodic memory loss—the inability to form new memories or recall recent events. In both diseases, a person might have a conversation, then forget it happened minutes later. Both diseases damage the hippocampus and surrounding medial temporal lobe structures, visible as atrophy on brain MRI. Both progress insidiously over years, and both disproportionately affect people over 80. For a clinician or family member observing the person’s day-to-day life, the presentation is nearly indistinguishable. The key difference lies in the speed and trajectory of that decline.
LATE patients show slower memory decline than patients with Alzheimer’s disease alone—a subtlety that only becomes apparent over months or years of observation. However, when LATE and Alzheimer’s co-occur, which happens in roughly 50 percent of Alzheimer’s patients, the disease course becomes significantly faster and more aggressive. A person with both pathologies deteriorates faster than neuropathology alone would predict. This is why diagnosis matters: if a patient is declining rapidly, the doctor may be looking at mixed pathology rather than pure Alzheimer’s, which changes prognostic expectations and treatment planning. The clinical presentation is further complicated by the fact that both diseases can occur in the same person without either being obvious. A patient might have amyloid plaques filling the brain (Alzheimer’s pathology) while TDP-43 accumulates separately in the hippocampus (LATE pathology), and cognitive testing alone cannot reveal which component is driving the memory loss most prominently.

The Biological Distinction—TDP-43 vs. Amyloid-Beta and Tau
LATE is caused by accumulation of TDP-43, a protein that in healthy brains regulates RNA and has normal cellular functions. In LATE, TDP-43 misfolds and aggregates, concentrating specifically in limbic structures—the hippocampus and amygdala—which are the brain’s memory and emotion centers. This selective, anatomically focused pathology is what gives LATE its clinical character: memory loss tends to be the dominant symptom, while other cognitive domains may remain relatively intact longer than in typical Alzheimer’s disease. Alzheimer’s disease, by contrast, is driven by accumulation of amyloid-beta (which forms plaques between neurons) and tau (which tangles inside neurons). These pathologies tend to spread more diffusely throughout the cortex, affecting not just memory but also language, executive function, and visuospatial abilities.
The topographical difference in brain pathology translates to differences in cognitive symptoms—though both diseases cause dementia, the cognitive profile often differs between pure LATE and pure Alzheimer’s. The clinical implication is crucial: a drug that clears amyloid-beta from the brain does nothing to a TDP-43 tangle or aggregate. If a person has pure LATE (TDP-43 pathology without significant Alzheimer’s pathology), anti-amyloid drugs like lecanemab and aducanumab are completely ineffective. This is not a failure of the drug; it is a mismatch between the pathology and the target. Conversely, if a person has mixed pathology—both amyloid and TDP-43—the picture becomes more complex. Anti-amyloid drugs may slow some aspects of cognitive decline driven by the Alzheimer’s component, but they leave the TDP-43-driven damage untouched.
Medial Temporal Lobe Atrophy—The Imaging Clue
Severe atrophy of the medial temporal lobe structures—specifically the hippocampus and amygdala—is a hallmark finding on brain MRI in LATE patients. This is not pathognomonic (unique to LATE); Alzheimer’s disease can also shrink these structures. However, the pattern and degree of atrophy can provide a clue to the underlying pathology. LATE tends to produce a more selective, severe pattern of hippocampal and amygdala shrinkage, while Alzheimer’s often produces more diffuse cortical atrophy affecting multiple brain regions. A radiologist or neurologist reviewing an MRI might note that a patient has profound medial temporal lobe atrophy with relatively preserved cortex elsewhere, which raises suspicion for LATE rather than typical Alzheimer’s pathology. This imaging pattern, combined with cognitive testing focused on episodic memory, becomes one piece of the diagnostic puzzle.
However, imaging alone cannot confirm LATE—it can only suggest the diagnosis and prompt further evaluation. The limitation to remember is that medial temporal lobe atrophy exists on a spectrum and overlaps between diseases. A person with pure Alzheimer’s disease can have significant hippocampal atrophy. Conversely, someone with LATE may have atrophy that is less pronounced than expected if the disease is early or progressing slowly. MRI is therefore a supporting diagnostic tool, not a definitive one. The formal diagnostic criteria, updated in 2025, require integration of cognitive history, imaging, and biomarkers to reach a diagnosis.

Why Current Alzheimer’s Medications Don’t Work for LATE
Lecanemab (marketed as Leqembi) and aducanumab (marketed as Aduhelm) are the first medications in decades to show slowing of cognitive decline in Alzheimer’s disease. Both are monoclonal antibodies that clear amyloid-beta from the brain. For people with Alzheimer’s disease driven by amyloid pathology, these medications can modestly slow progression—though the benefit is modest, typically extending the rate of decline by several months over a few years. For people with LATE, these anti-amyloid medications are ineffective. Because LATE is driven by TDP-43 accumulation, not amyloid-beta, clearing amyloid from the brain does not address the underlying pathology.
Giving lecanemab to someone with pure LATE is similar to prescribing a blood pressure medication to someone whose problem is diabetes; the drug works perfectly in its intended target, but it addresses the wrong disease. The clinical complexity arises when a patient has mixed pathology—both Alzheimer’s and LATE. In such cases, an anti-amyloid drug might slow the Alzheimer’s component of cognitive decline while leaving the LATE component untouched. A patient taking lecanemab might decline more slowly than without it, but the overall trajectory remains steeper than in someone with Alzheimer’s alone, because the TDP-43 pathology continues unchecked. Families hoping for a medication to “fix” the dementia must understand that available medications address only part of the problem in mixed pathology, and address none of it in pure LATE.
Gene Therapy and Precision TDP-43 Treatments on the Horizon
Because LATE is a relatively newly recognized disease entity—it was formally distinguished from Alzheimer’s only in the past 15 years—development of LATE-specific treatments lags behind Alzheimer’s research. However, the landscape is shifting. NIH-funded researchers have developed gene therapy approaches that target TDP-43 specifically. These experimental treatments take a precision medicine approach: rather than broadly suppressing TDP-43 (which would be harmful, since the protein has normal cellular functions), the therapy activates only in neurons that already have dysfunctional, misfolded TDP-43. The concept is elegant: the gene therapy can identify which neurons are sick and selectively correct the problem in those cells without disrupting healthy neurons where TDP-43 is functioning normally. This precision approach represents a major shift away from the blunt-instrument strategies of current Alzheimer’s treatments.
Beyond gene therapy, emerging research focuses on vascular stability, glial cell function (microglia and astrocytes are implicated in TDP-43 pathology), and direct TDP-43 aggregation inhibitors. These therapies are still in development and not yet available to patients outside clinical trials, but they signal the direction of future treatment. The current reality is that there is no approved medication that specifically targets TDP-43. Patients with LATE diagnosed today have no disease-modifying treatment tailored to their pathology. Symptom management—addressing behavioral issues, memory support, and quality of life—remains the mainstay of care. For families grappling with LATE, understanding that specific treatments are in development but not yet available helps set realistic expectations and may inform decisions about participation in clinical trials.

The 2025 Diagnostic Criteria—How Doctors Now Reliably Identify LATE
For decades, LATE could only be definitively diagnosed at autopsy when pathologists examined brain tissue under a microscope and found TDP-43 accumulation in the limbic system without significant amyloid or tau pathology. This autopsy-only diagnosis meant that during a patient’s lifetime, LATE could be suspected but never confirmed. In January 2025, this changed. New clinical diagnostic criteria for LATE were formally published and validated, allowing neurologists and dementia specialists to diagnose LATE with reasonable confidence during life. The 2025 criteria integrate cognitive history, structural brain imaging, cerebrospinal fluid (CSF) biomarkers, and PET imaging.
A diagnosis of “probable LATE” requires a pattern of progressive episodic memory loss, evidence of medial temporal lobe atrophy on MRI, amyloid-negative biomarkers (meaning amyloid-beta pathology is absent or minimal), and tau-negative or tau-minimal biomarkers. If biomarkers are unavailable or inconclusive, a diagnosis of “possible LATE” can be made based on cognitive presentation and imaging alone. These criteria do not require absolute certainty—they provide a probabilistic framework that recognizes the biological reality that most dementia involves mixed pathologies and biomarkers are imperfect measures. The practical implication is that a person presenting with progressive memory loss and hippocampal atrophy can now be evaluated with biomarker testing (blood tests or CSF studies) and brain imaging to determine whether amyloid and tau are present. If they are absent or minimal, and TDP-43 is elevated in biomarkers, probable LATE can be diagnosed. This allows for more accurate prognostication, better-informed family discussions, and potentially better matching of patients to clinical trials of TDP-43-targeted therapies when they become available.
The Prevalence Epidemic—Why LATE Is Far More Common Than Most People Realize
Among people aged 85 and older, approximately one in three has LATE. In the 90+ age group, prevalence rises to one in three or higher. These are staggering numbers, yet LATE remains largely unknown among the general public and many primary care physicians. The reason is partly historical: LATE was not formally characterized until 2011 and did not gain widespread clinical attention until several years later. Many older adults with LATE were diagnosed with Alzheimer’s disease instead, either because that was all the diagnostic tools could confirm at the time, or because true Alzheimer’s pathology was also present.
The co-pathology picture is equally striking. Approximately 50 percent of people with Alzheimer’s disease neuropathology also have concurrent LATE pathology. This means that half of dementia patients with confirmed amyloid and tau also have TDP-43 accumulation. For clinical practice, this translates to a substantial proportion of dementia patients who would not fully respond to anti-amyloid drugs, because their cognitive decline is driven partly by TDP-43 pathology untouched by those medications. As the U.S. population continues to age and more people reach the 85+ age group, the absolute number of people with LATE will increase substantially, making diagnosis and differentiation from Alzheimer’s increasingly important for clinical practice.
Conclusion
LATE dementia and Alzheimer’s disease are distinct biological entities that share superficial similarities in clinical presentation but require fundamentally different treatment approaches. Both cause progressive memory loss anchored in medial temporal lobe damage, and both strike primarily in the oldest age groups. However, LATE is driven by TDP-43 pathology localized to limbic structures, while Alzheimer’s is driven by amyloid-beta and tau spreading diffusely through the cortex. Current anti-amyloid medications effectively target Alzheimer’s pathology but are completely ineffective for LATE.
As gene therapy approaches and other TDP-43-specific treatments emerge from research pipelines, the importance of accurate diagnosis becomes clear: a patient with pure LATE given only anti-amyloid drugs will not benefit from treatments that target the wrong pathology. For patients and families, the path forward involves three key actions. First, understand that memory loss in an older adult warrants evaluation for the underlying pathology driving it—Alzheimer’s, LATE, mixed pathology, or one of several other possible causes. Second, advocate for biomarker testing and structural imaging if a dementia diagnosis is made; the new 2025 criteria provide a framework for accurate LATE diagnosis that was unavailable just months ago. Third, stay informed about emerging LATE-specific treatments and consider clinical trial participation if appropriate, because the next generation of dementia medications will be tailored to specific pathologies rather than administered based on symptoms alone.
You Might Also Like
- What Is LATE Dementia and Why Neurologists Say It Is the Most Misdiagnosed Form
- What Is LATE Dementia and Why Neurologists Say It Is the Most Misdiagnosed Form
- How Lewy Body Dementia Differs From Alzheimer’s and Why Getting the Right Diagnosis Matters
For more, see NIH MedlinePlus — dementia.